What's hidden in your GWAS?
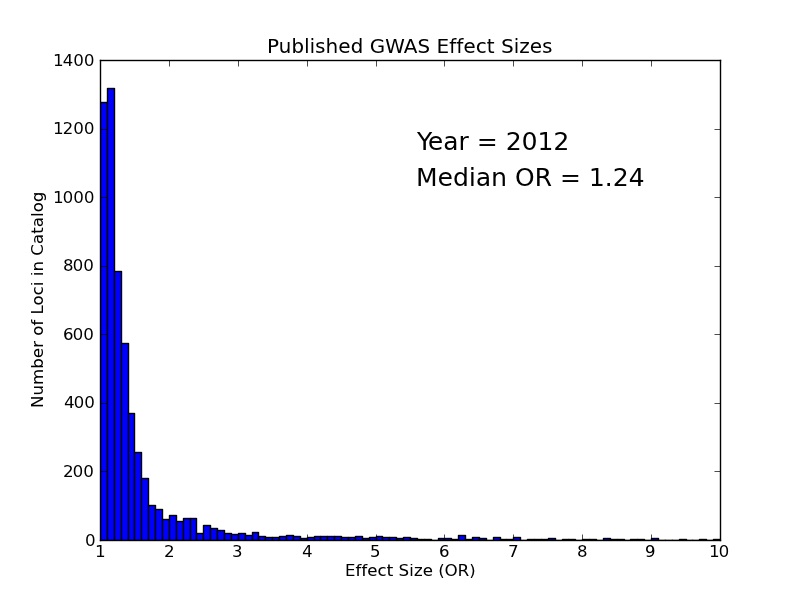
Over the past decade, genome-wide association studies (GWAS) have identified numerous loci for a range of complex diseases. Often these genomic locations fall within non-coding regions that have limited annotation and no obvious functional consequence. Additionally, most loci account for only a small fraction of disease heritability, with the vast majority of published SNPs having odds ratios under two. The standard GWAS analysis paradigm is ill-suited for collecting these functionally ambiguous, small-effect associations and placing them in an informative context. Given the apparent polygenic nature of most complex disease, better integrative approaches are required to extract information from all disease associations, not just the top hits, and place them in an informative context.
|
How can Sherlock help? |
|

How do I use Sherlock?
Simply submit your list of GWAS associations (SNPs and p-values) as described here. It is important to upload all SNPs in your association study, not just the top hits. Sherlock may be able to group multiple lower-confidence SNPs to discover functionally-important genes. The system will email results to you, usually within one day. Currently, all eQTL studies in our database are for CEU cohorts. If you have access to imputed SNPs (not just the set of SNPs on your microarray), please use these.
|
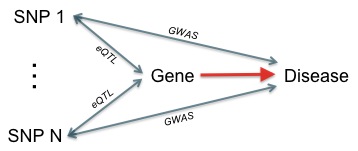
How does it work? |
|
{kind=link}