GWAS for Complex Disease
Genome wide association studies (GWAS) for complex disease typically yield a large number of loci, often in non-coding regions, with limited annotation and no obvious functional consequence. Additionally, many more loci fail to reach genome-wide significance in a single study but are clearly in excess relative to expectations under the null distribution. Together, such limitations may frustrate efforts to understand complex disease etiology, since statistically-significant, well-annotated loci usually explain only a small fraction of disease heritability.
What is needed are straightforward methods to mine weaker GWAS associations and place large numbers of loci in the context of functionally-relevant genes and gene networks. Sherlock attempts to do this by scoring GWAS results against thousands of genes from expression quantitative trait loci (eQTL) studies. A strong body of evidence suggests that changes in gene expression play a key role in complex disease. Genes that share associations at multiple loci with the GWAS are unexpected by chance and, thus, may play a causal role in the disease. In our testing, many of the supporting loci for candidate genes are in trans (i.e. distal to the gene itself), suggesting that this approach may be particularly useful in understanding the role of gene regulation in disease.
The Importance of Distal (trans) Genomic Loci
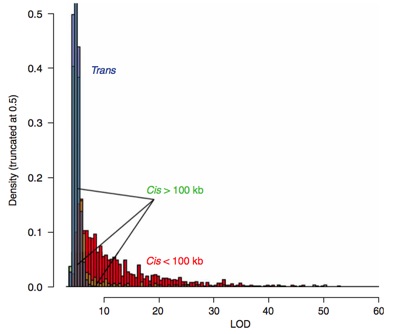
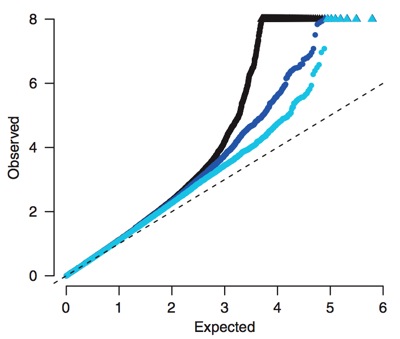
Both theoretical and observational studies indicate that evolution can yield complex, highly-redundant networks of gene regulation, most likely to maintain robust control in varying environments [Soyer 2006]. Our understanding of the role that distal loci play in these networks is limited, but recent genome wide characterizations of open chromatin, nucleosome positioning, and transcription factor binding underscore their importance. Regulatory elements far outnumber genes, with most distal to the genes that they control [ENCODE 2007, ENCODE 2012]. Although individual proximal elements typically have a stronger influence on gene expression (figure below left), a large number of distal loci may operate in concert to influence a gene’s transcript level. In the context of GWAS, distal elements are critical to understanding the residual associations not explained by either the significant loci or population artifacts. Typically, only part of the execess associations (relative to expectations under the null) in a given GWAS can be explained by loci proximal to established disease genes (figure below right).
 |
 |
Sherlock Genetic Signature Matching
Sherlock is based on the idea that SNPs associated with the expression level of a given gene (or that of any quantitative molecular trait) can be used to mine GWAS results for insight regarding gene-disease associations. A gene that is causal for the disease may have multiple expression SNPs (eSNPs). Genotype polymorphisms at these eSNPs can alter gene expression, which may in turn alter the disease risk. Therefore, many of the gene's eSNPs are likely to be associated with the disease as well. In general, significant overlap of the eQTL of a gene and the loci associated with the disease would imply a likely functional role for the gene in the disease.
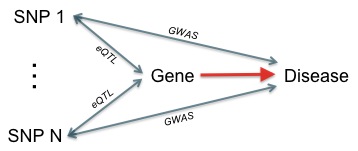 |
Using gene-specific "genetic signatures" (i.e. patterns of associations) in eQTL data, it is conceptually straightforward to test the alignment of a given gene against GWAS (figure below). In practice, the analysis is complicated by linkage patterns, wide variation in the number of eQTL per gene, the non-linear nature of the input p-values, and other issues.
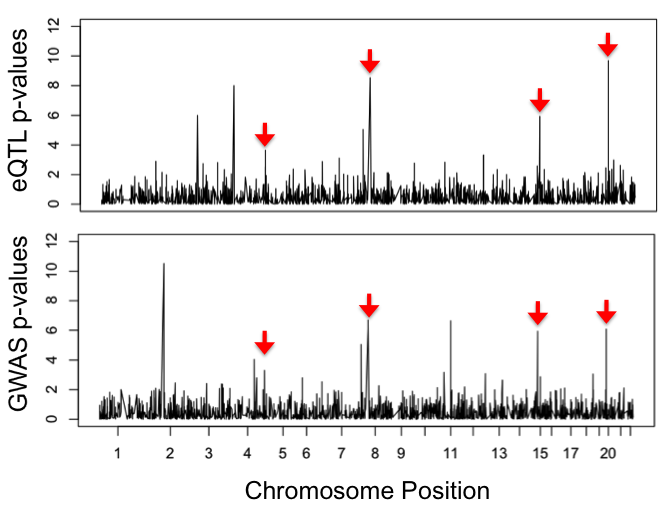 |
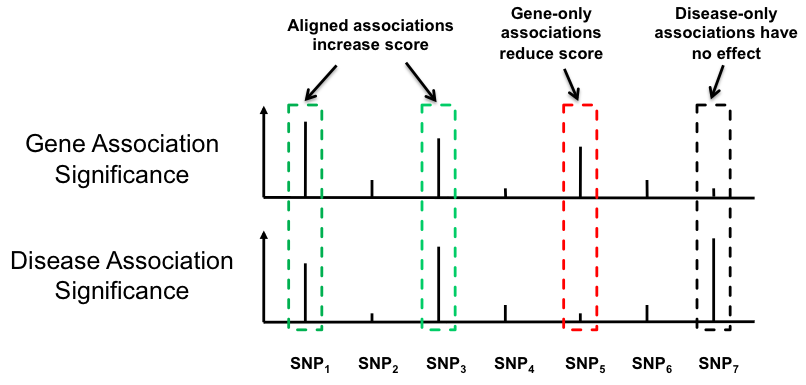
Sherlock's scoring rubric increases the total gene score for overlapping SNPs (e.g SNPs 1 and 3, in green, in the figure below) and provides a penalty in the absence of overlap (SNP 5, in red). Associations found only in the GWAS (SNP 7, in black) do not alter the score. Sherlock computes individual Log Bayes Factors (LBFs) for each SNP pair in the alignment; the sum of these constitutes the final LBF score for each gene. Sherlock also computes p-values for each gene score via simulation. A gene is considered significant when its p-value exceeds a certain threshold.
 |
Complete details are provided in our manuscript [He 2013].